With the following input parameters for the atompaw code:
|
PAW Trial
|
Exc. Form
|
Rc (bohr)
|
atompaw Input File
|
atompaw Output File
|
|
#1
|
GGA
|
1.0
|
||
|
#1
|
LDA
|
1.0
|
||
|
#2
|
GGA
|
1.41
|
||
|
#2
|
LDA
|
1.41
|
The pwpaw code was used to make the following crystals yielding the following output properties:
|
Crystal (type)
|
Test Results
|
Convergence Graphs
|
Sample pwpaw Input File
|
Sample pwpaw Output File
|
|
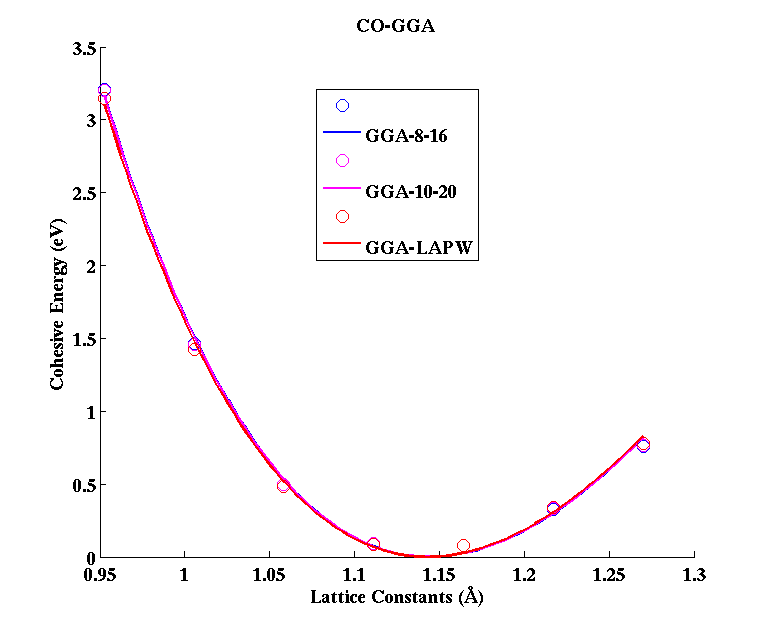
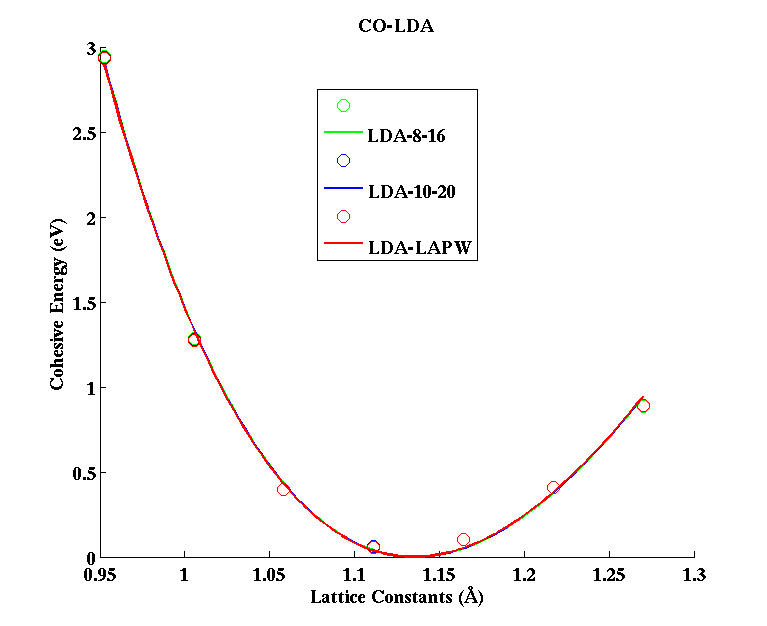
CO (diatomic molecule)
|
(PAW #1)
|
|||
|
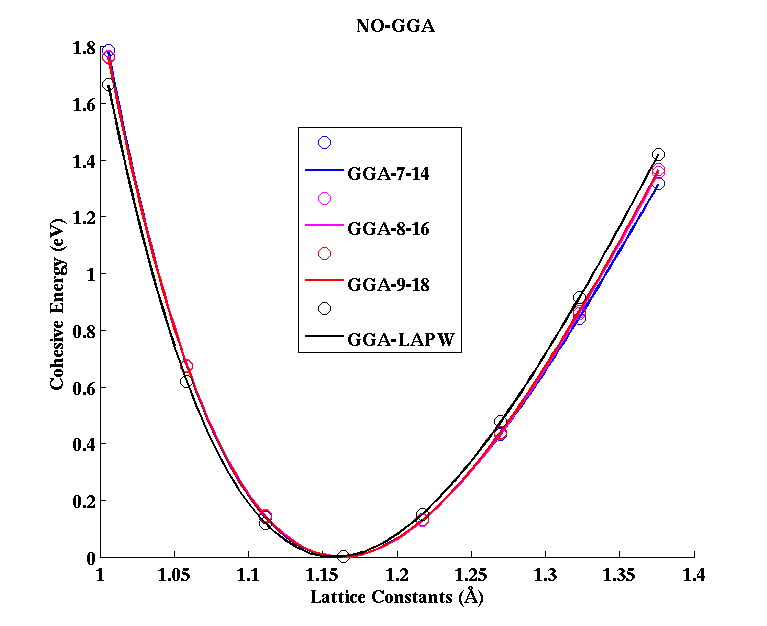
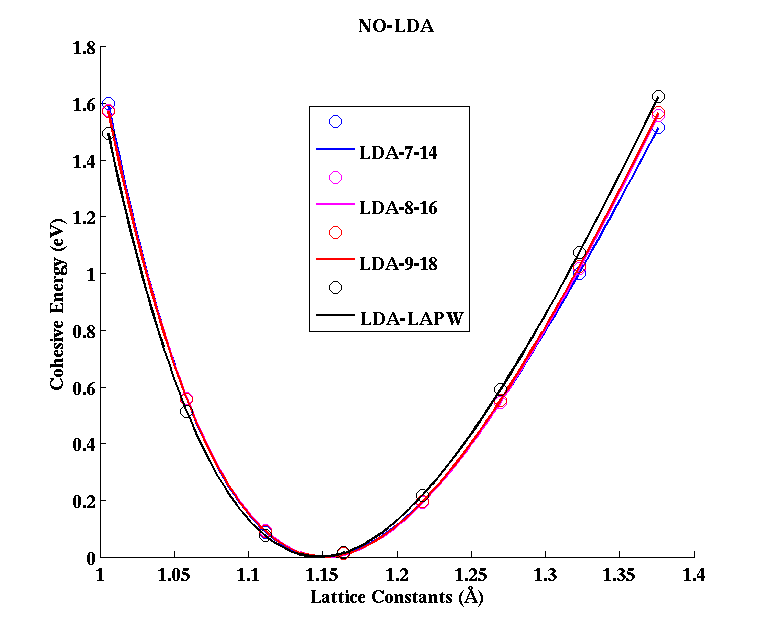
NO (diatomic molecule)
|
(PAW #1)
|
|||
|
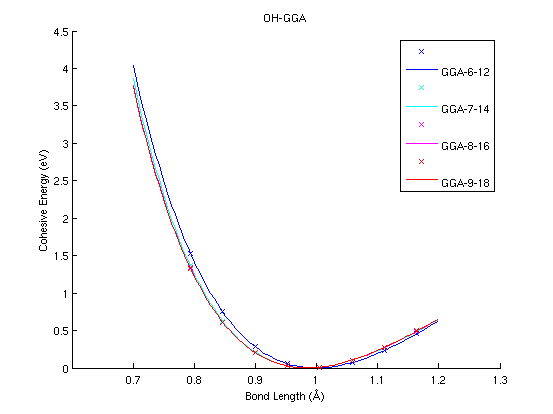
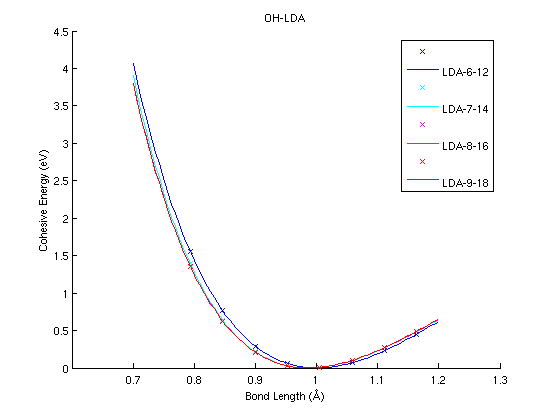
OH (diatomic molecule)
|
(PAW #1)
|
|||
|
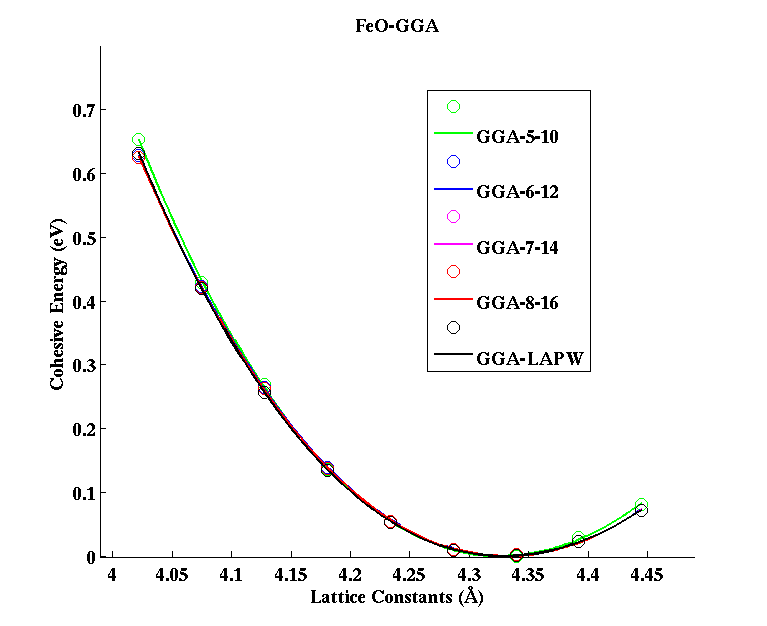
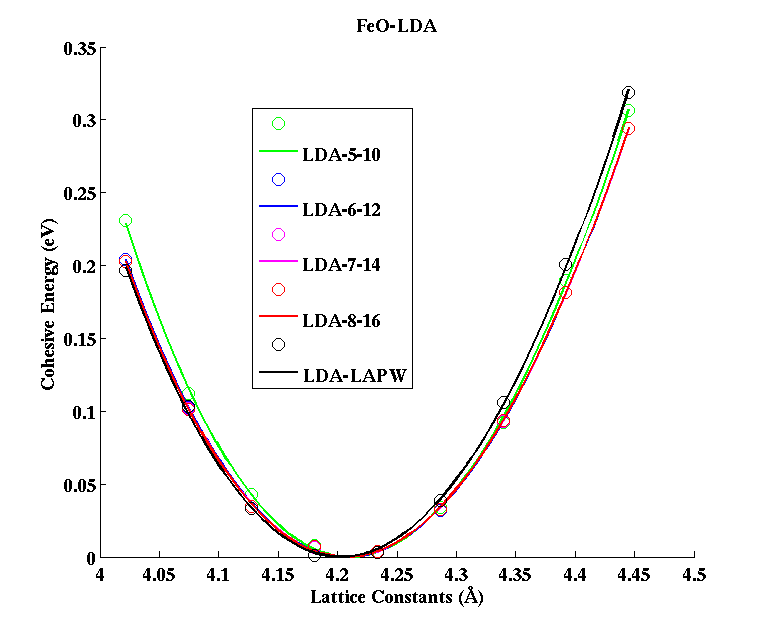
FeO (FCC)
|
(PAW #2)
| |||
|
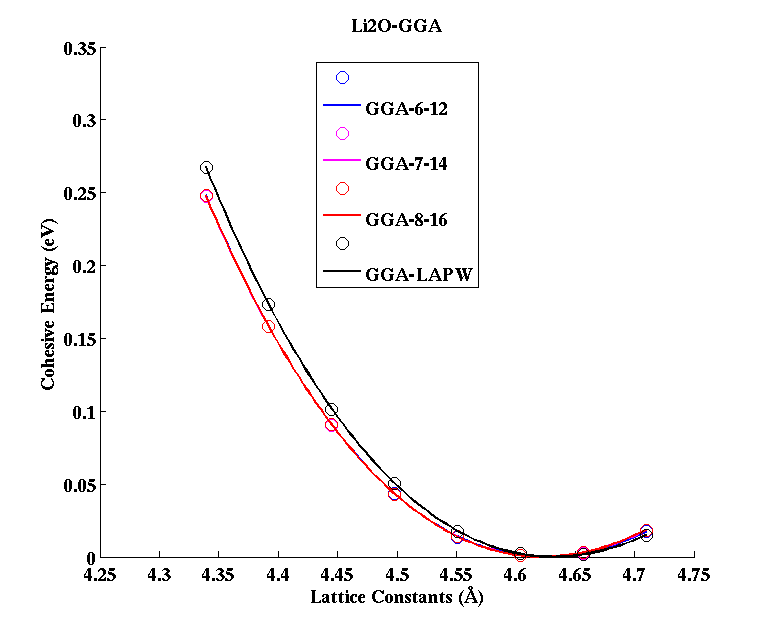
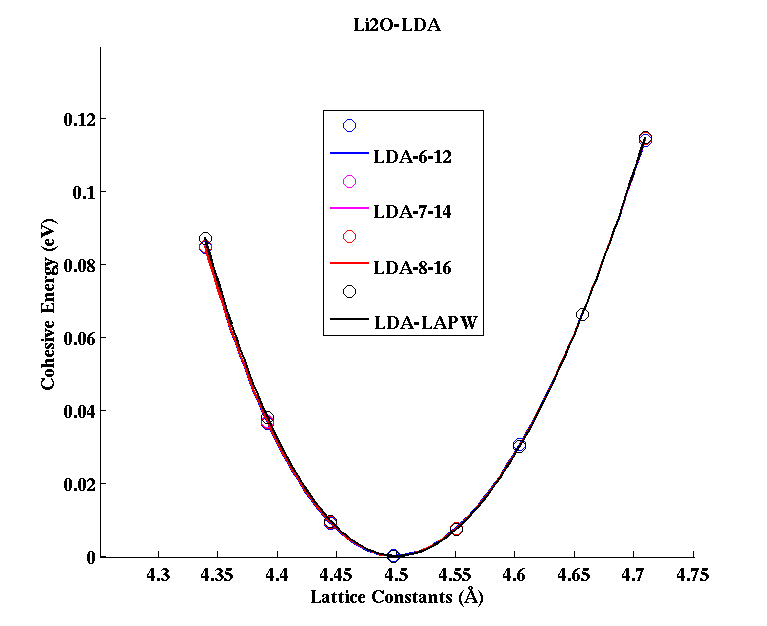
Li2O (FCC)
|
(PAW #2)
| |||
|
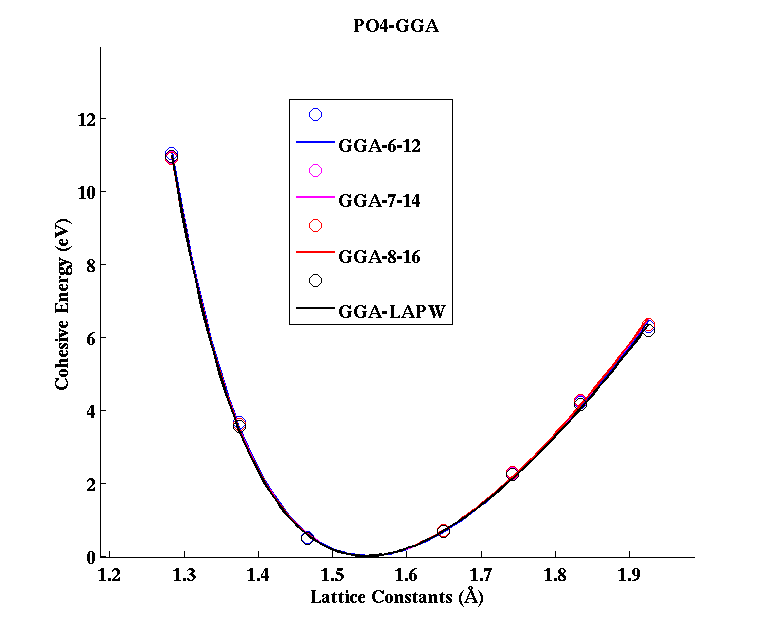
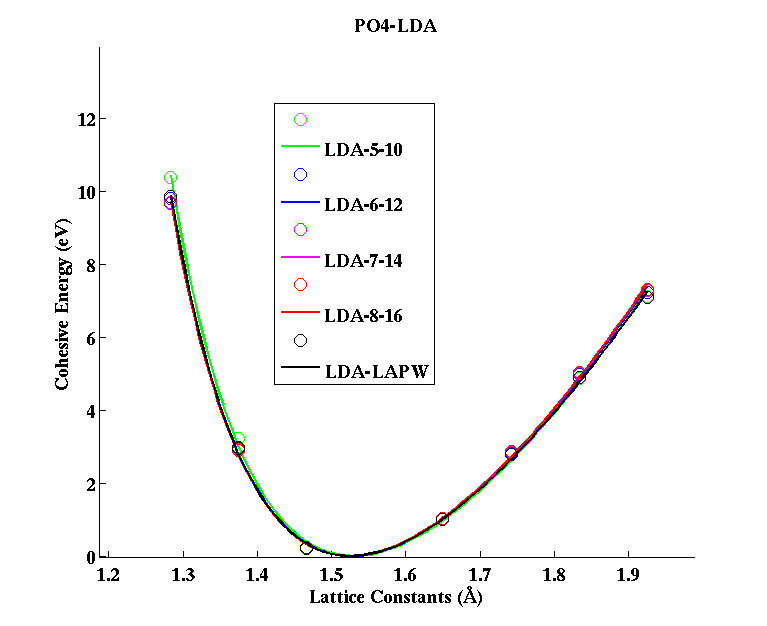
PO4 (tetrahedral molecule)
|
(PAW #2)
| |||
|
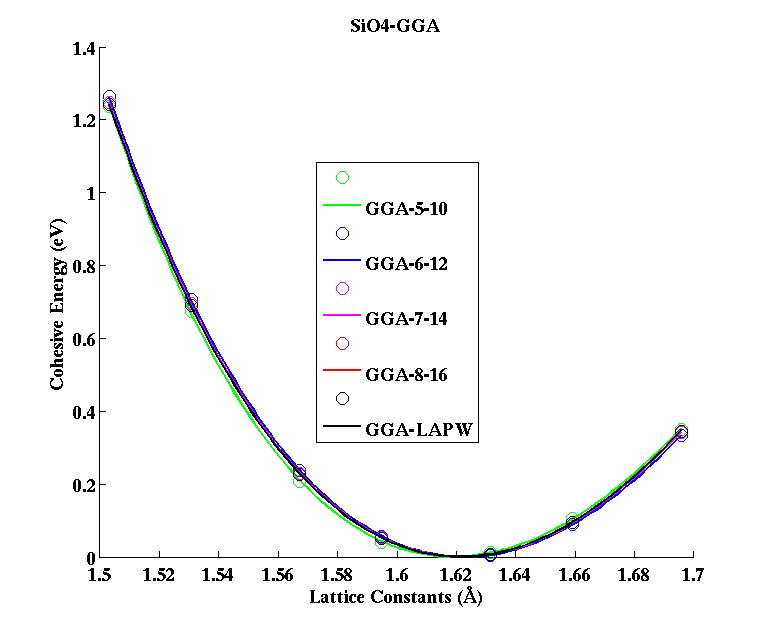
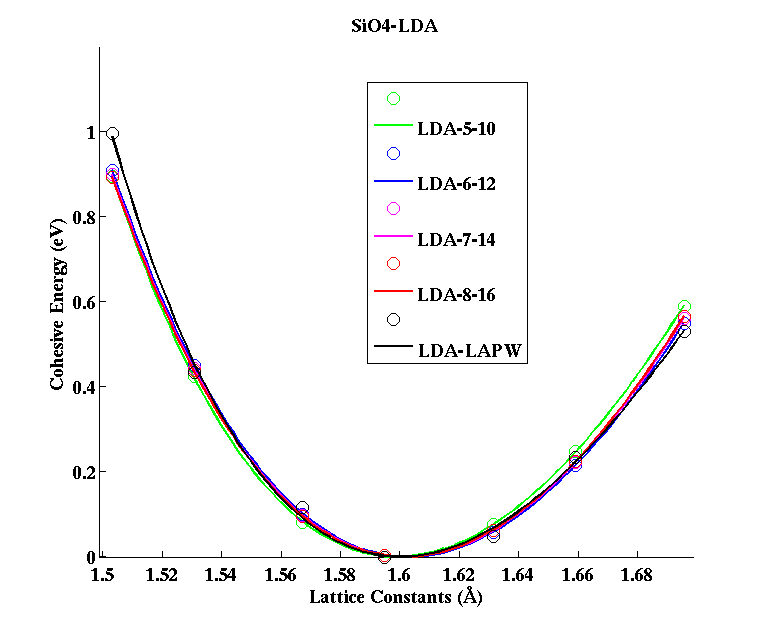
SiO4 (tetrahedral molecule)
|
(PAW #2)
| |||
|
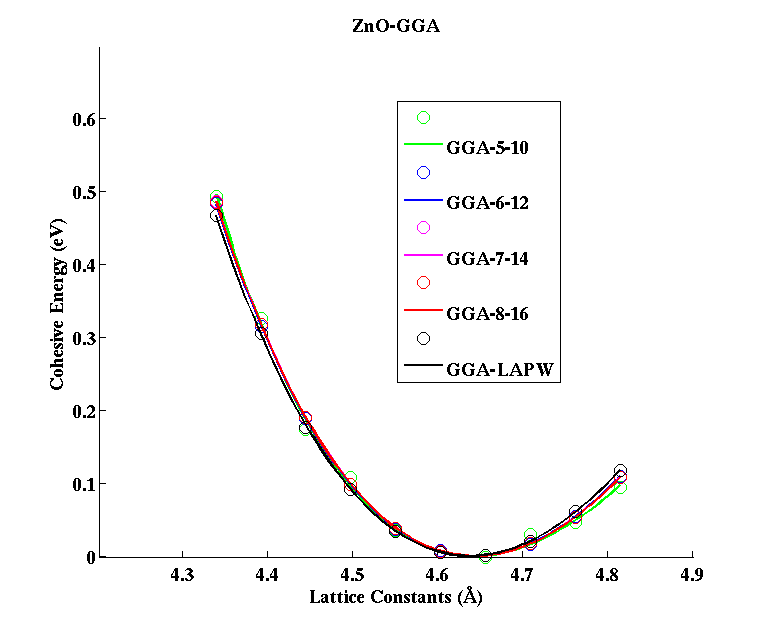
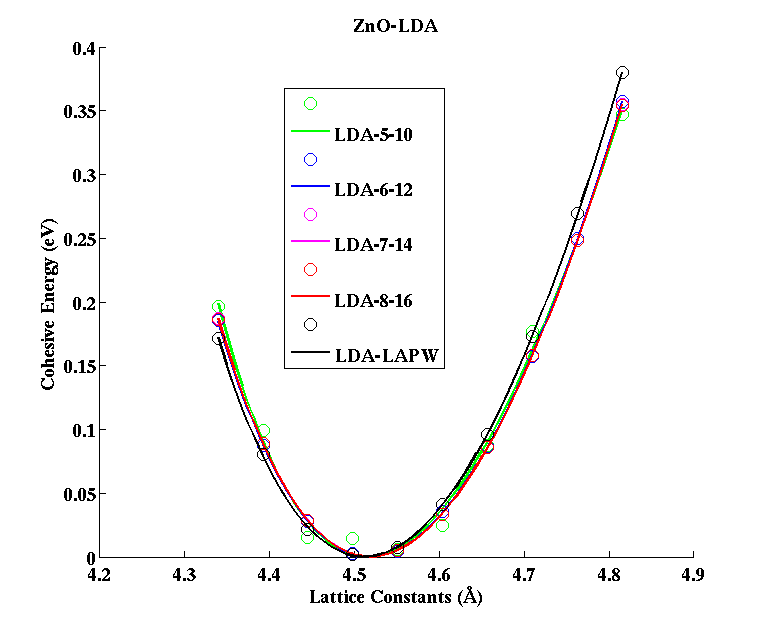
ZnO (Zincblende)
|
(PAW #2)
| |||
|
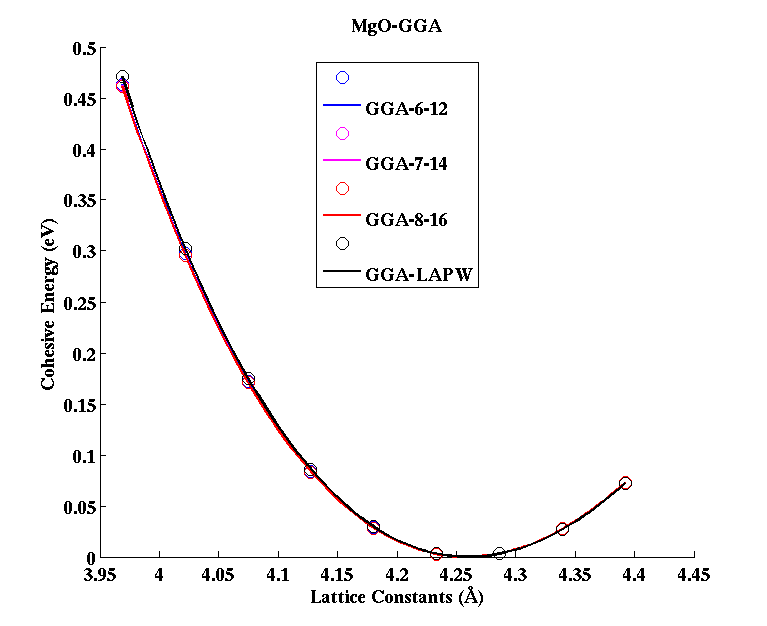
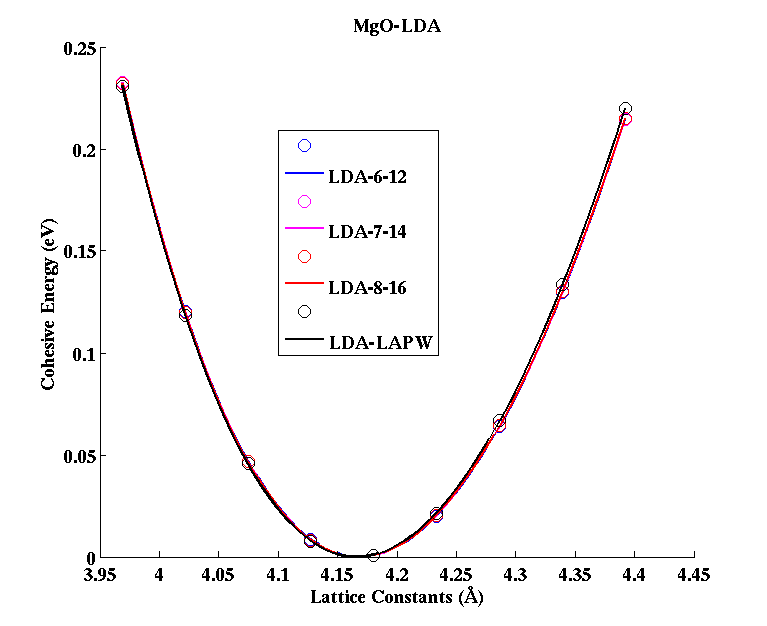
MgO (FCC)
|
(PAW #2)
| |||
|
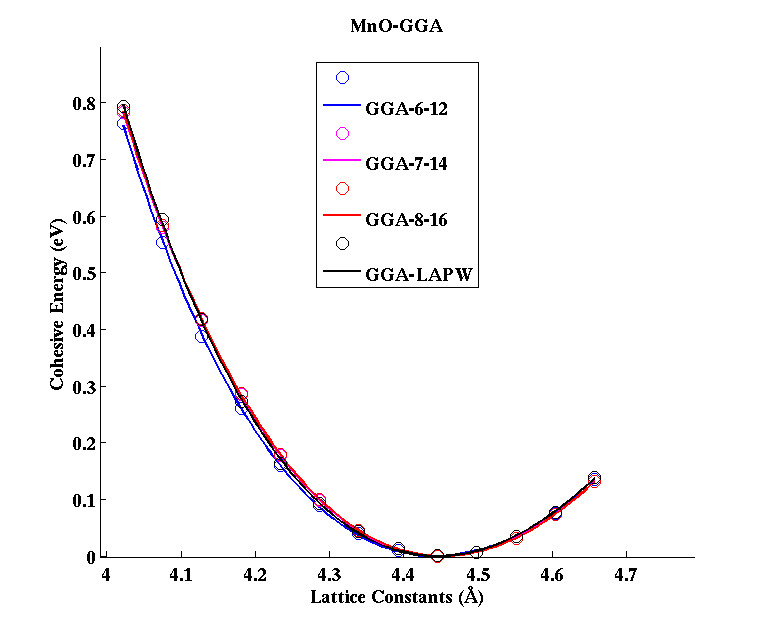
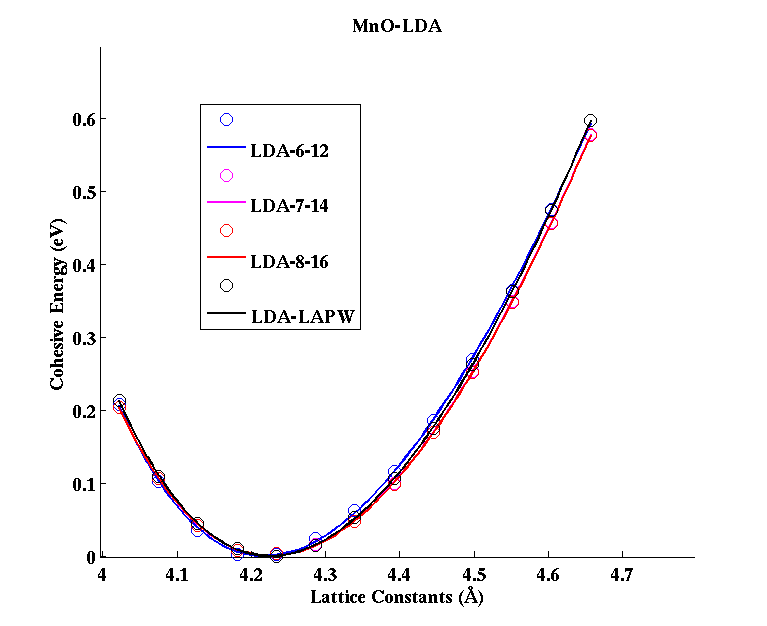
MnO (FCC)
|
(PAW #2)
|
|||
|
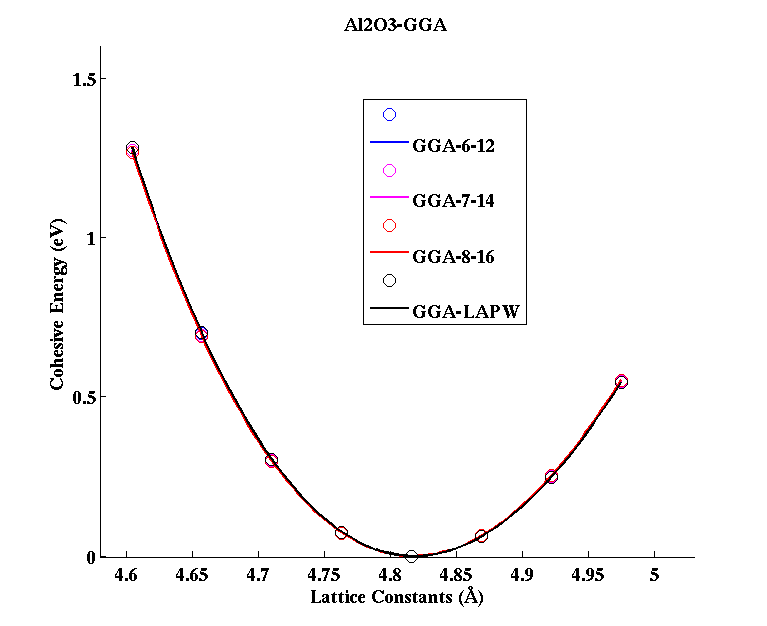
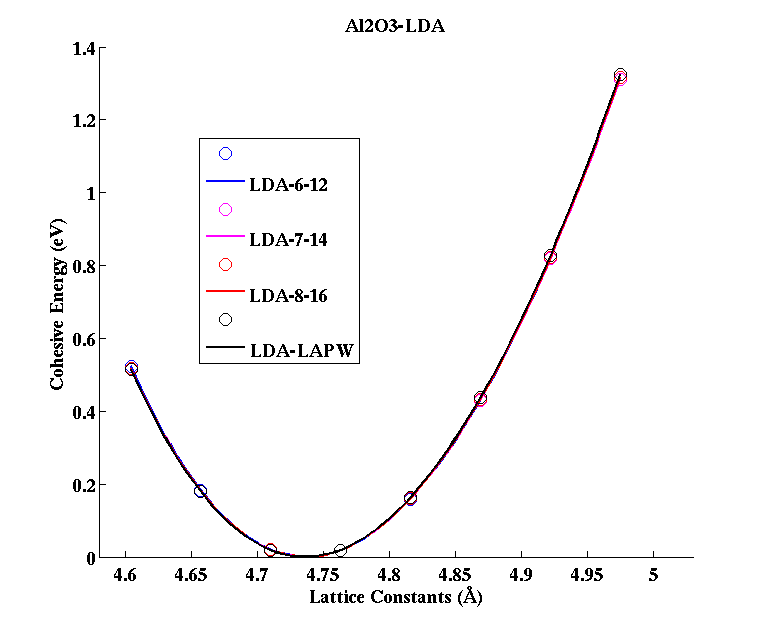
Al2O3 (Corundum)
|
(PAW #2)
|
|||
|
Crystal 12
|
-
|
-
|
-
|
-
|
This page last modified on July 30, 2008 by David Harris
Please send questions or comments to David Harris at harrisdt@wfu.edu or N.A.W. Holzwarth at natalie@wfu.edu
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}