With the following input parameters for the atompaw code:
|
PAW Trial
|
Core States
|
Valence States
|
Rc (bohr)
|
Exc. Form
|
Additional Functions?
|
atompaw Input File
|
atompaw Output File
|
|
#1
|
1s, 2s, 3s, 4s, 2p, 3p, 4p, 3d
|
5s, 5p, 4d
|
3.35
|
LDA-PW
|
No
|
||
|
#2
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
#3
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
The pwpaw code was used to make the following crystals yielding the following output properties:
|
Crystal (type)
|
Lattice Constant (Å)
|
Bulk Modulus (GPa)
|
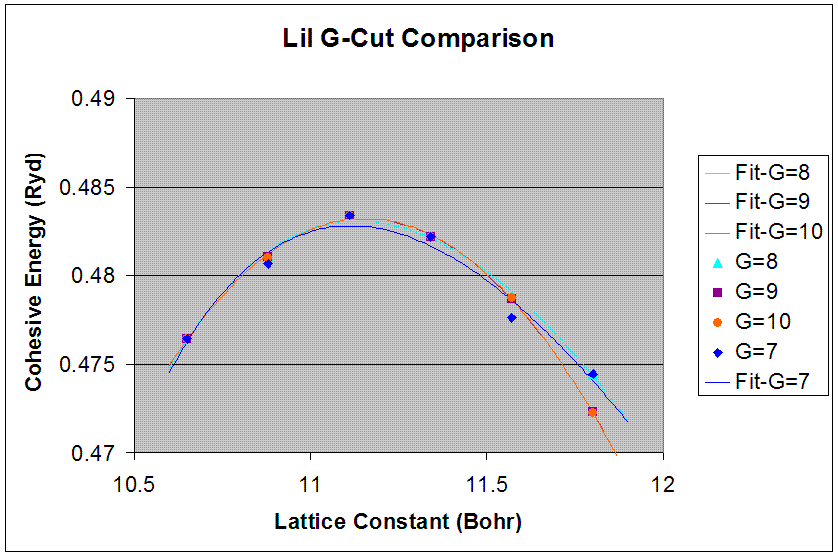
Appropriate G-Cut
|
pwpaw Input File
|
pwpaw Output File
|
|
LiI (FCC)
|
5.90 (PAW #1) 6.00 (expt. ref 3) |
32.1 (PAW #1) |
|||
|
Crystal 2
|
- |
- |
-
|
- |
-
|
|
Crystal 3
|
-
|
-
|
-
|
-
|
-
|
The pwpaw input and output files correspond to a g-cut of 8 for the smallest lattice constants of the six data points. The given k-points and crystal-symmetry input files were used for each lattice constant data point and g-cut trial for each crystal.
This page last modified on July 26, 2005 by John Tumbleston
Please send questions or comments to John Tumbleston at jtumbleston@elon.edu or N.A.W. Holzwarth at natalie@wfu.edu
{kind=link}