With the following input parameters for the atompaw code:
|
PAW Trial
|
Core States
|
Valence States
|
Rc (bohr)
|
Exc. Form
|
Additional Functions?
|
atompaw Input File
|
atompaw Output File
|
|
#1
|
1s, 2s, 3s, 2p, 3p, 3d
|
4s, 5s, 4p, 5p, 4d
|
2.6
|
LDA-PW
|
No
|
||
|
#2
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
|
#3
|
-
|
-
|
-
|
-
|
-
|
-
|
-
|
The pwpaw code was used to make the following crystals yielding the following output properties:
|
Crystal (type)
|
Lattice Constant (Å)
|
Bulk Modulus (GPa)
|
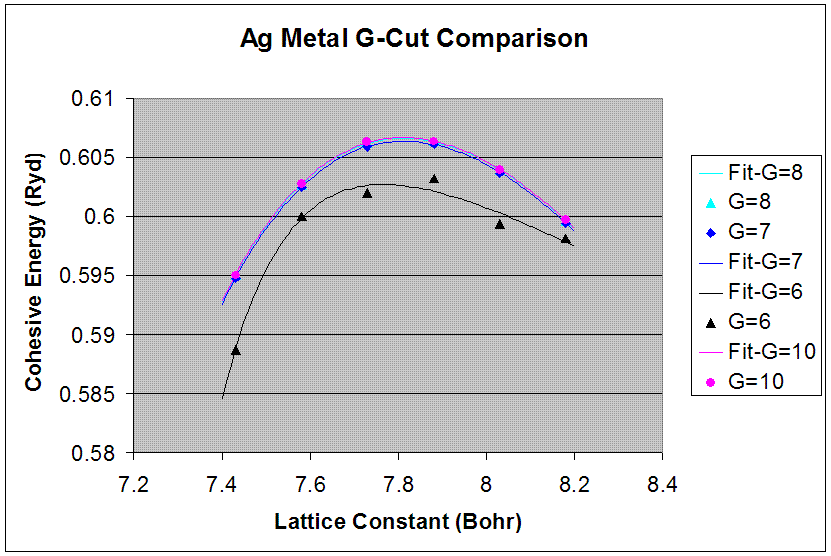
Appropriate G-Cut
|
pwpaw Input File
|
pwpaw Output File
|
Spin-up minus Spin-down value
|
|
Ag Metal (FCC)
|
4.13 (PAW #1) 4.00 (LSDA theory ref 1) 4.07 (expt. ref 1) |
104. (PAW #1) 149. (LSDA theory ref 1) 109. (expt. ref 1) |
0
|
|||
|
Crystal 2
|
-
|
-
|
-
|
-
|
-
|
-
|
|
Crystal 3
|
-
|
-
|
-
|
-
|
-
|
-
|
The pwpaw input and output files correspond to a g-cut of 8 for the smallest lattice constants of the six data points. The given k-points and crystal-symmetry input files were used for each lattice constant data point and g-cut trial for each crystal. The "spin-up minus spin-down" value corresponds to a g-cut of 8 for the lattice constant that is closest to the maximum energy value.
This page last modified on July 26, 2005 by John Tumbleston
Please send questions or comments to John Tumbleston at jtumbleston@elon.edu or N.A.W. Holzwarth at natalie@wfu.edu
{kind=link}