Current Research
Macosko Lab
There will be more information added later with our current research. For the time being, the homepage describes some of our past research questions.
Past Research
Studies of vesicle transport by kinesin in living neurons
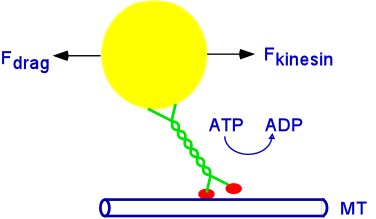
Until recently our research project has been an extension of Professor G. Holzwarth's work: the measurements of the drag force and mechanical work required for fast transport of vesicles and the relationship of this cellular task to the known limitations of motor proteins, especially kinesin. In buffer, against the force of an optical trap, the maximum of steady force which kinesin can exert is 6.5 pN. One ATP is hydrolyzed per 8 nm step, and each step takes 50 microseconds. About 100 such steps occur per second during processive movement. In a cell, the vesicle(load) is in cytoplasm, not buffer+trap, so the load on the motor is viscoelastic drag. The viscous part of this load differs by a factor of 10,000-100,000 from the viscous load in an optical trap, since the viscosity of water is .001 Pa*s and the zero-frequency viscosity of cytoplasm is roughly 50 Pa*s. Does kinesin develop the same force in these two environments? Our goal is to measure the forces and work required to move vesicles in live cells and to compare these to the limits established for kinesin in solution.
One expects highly extended cells such as neurons to be particularly sensitive to the energy costs of vesicle transport. For this reason, we are measuring vesicle transport in differentiated PC12 cells, which are a good model system, for neurons, but easier to grow.
Single-molecule fluorescence microscopy of actively transcribing T7 RNA polymerase
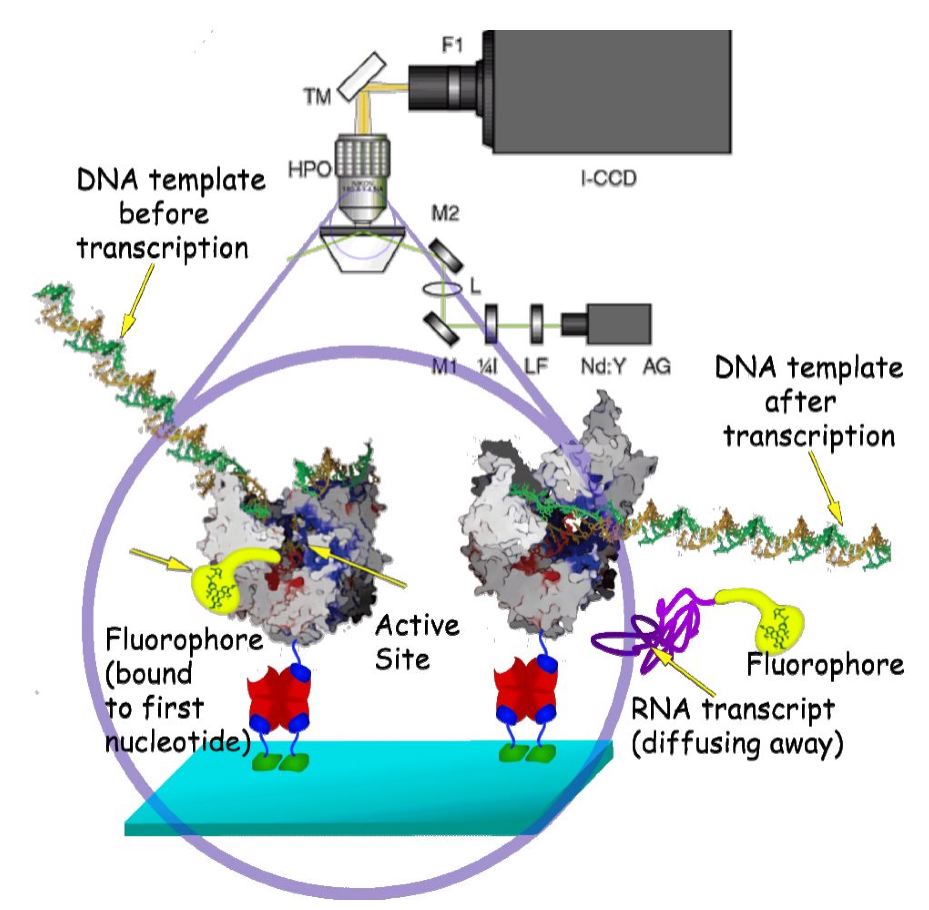
In the most basic T7 RNA polymerase (RNAP) experiment, a single fluorophore, covalently attached to GTP, will be incorporated at the 5' end of the RNA, thus marking the beginning of transcription (see figure). The signal from this incorporated fluorophore will persist until transcription terminates and the transcript diffuses away. By fitting histograms of the fluorescent persistence times, to kinetic models we can uncover essential information (average rate, processivity, abortive transcription percentage, etc.) regarding actively transcribing T7 RNAP.
Experiments to investigate gene expression and regulation at the single-molecule level
One of the simplest known gene regulation systems is the autoregulation of lysozyme in T7 bacteriophage. T7 lysozyme is produced during a T7 infection to help lyse the bacterial cell wall in order to release the newly formed bacteriophage capsids. Additionally, T7 lysozyme autoregulates by inhibiting transcription by T7 RNAP. Using single-molecule fluorescence, we will observe the processive transcription rate of single T7 RNAP's in the presence of T7 lysozyme. Putative sequence dependence of the autoregulatory effect will also be examined.
Single-molecule fluorescence resonance energy transfer analysis of transcription initiation
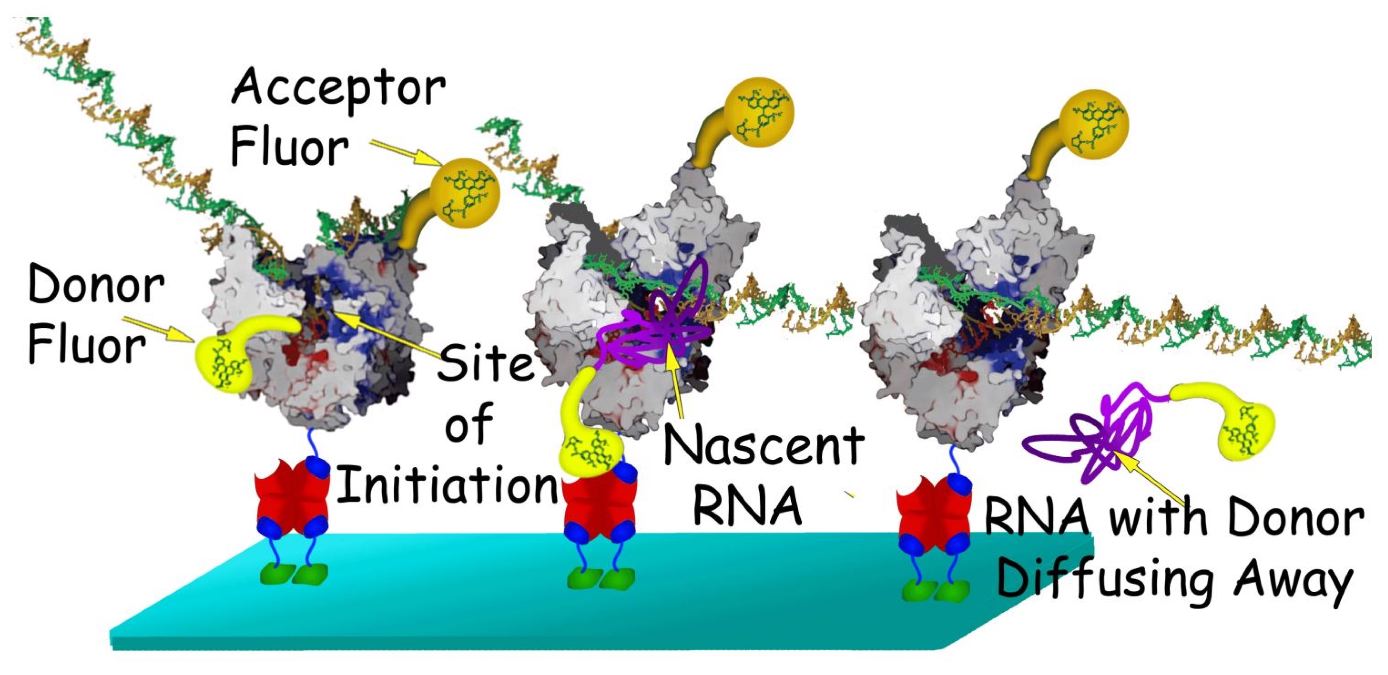
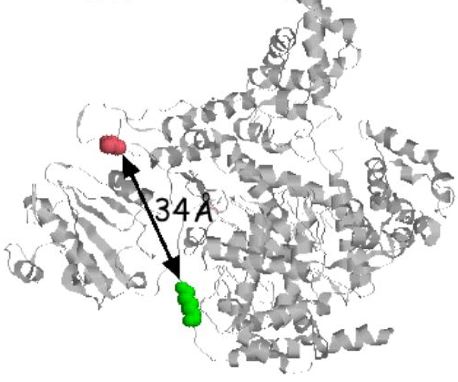
Single molecule FRET is a powerful tool to probe mechanical motion in protein machines. In the FRET experiment (see figure), donor-labeled GTP will associate with the template DNA at the first position of transcription. Meanwhile, a his-tag, genetically engineered at the N-terminus of recombinantly expressed T7 RNAP, will bind tightly to the acceptor fluorophore at the N-terminal end of each surface-immobilized polymerase. In this way fluorescent transfer between donor and acceptor will begin immediately upon transcription initiation. The fluorescent signal from the donor and the energy transfer to the acceptor will suddenly drop if RNA transcription is aborted. Since aborts are quite common, many of the data traces will last only a fraction of a second. These short traces will be useful in characterizing the transition from initiation to elongation. For example, in a single molecule study of the E. coli Rep helicase, researchers found that the FRET signal oscillated markedly when the helicase paused on the DNA. Accordingly, we will look for a characteristic FRET signal in the abortive data that will shed light on the failed transition to elongation. Longer traces will also be informative: their FRET signal intensity should abruptly decrease at the transition between initiation and elongation. We will correlate this abrupt change to the initiation and the elongation crystal structures and help clarify the mechanical motions of this important transition.
Integration of an atomic force microscope (AFM) with single molecule fluorescence microsopy
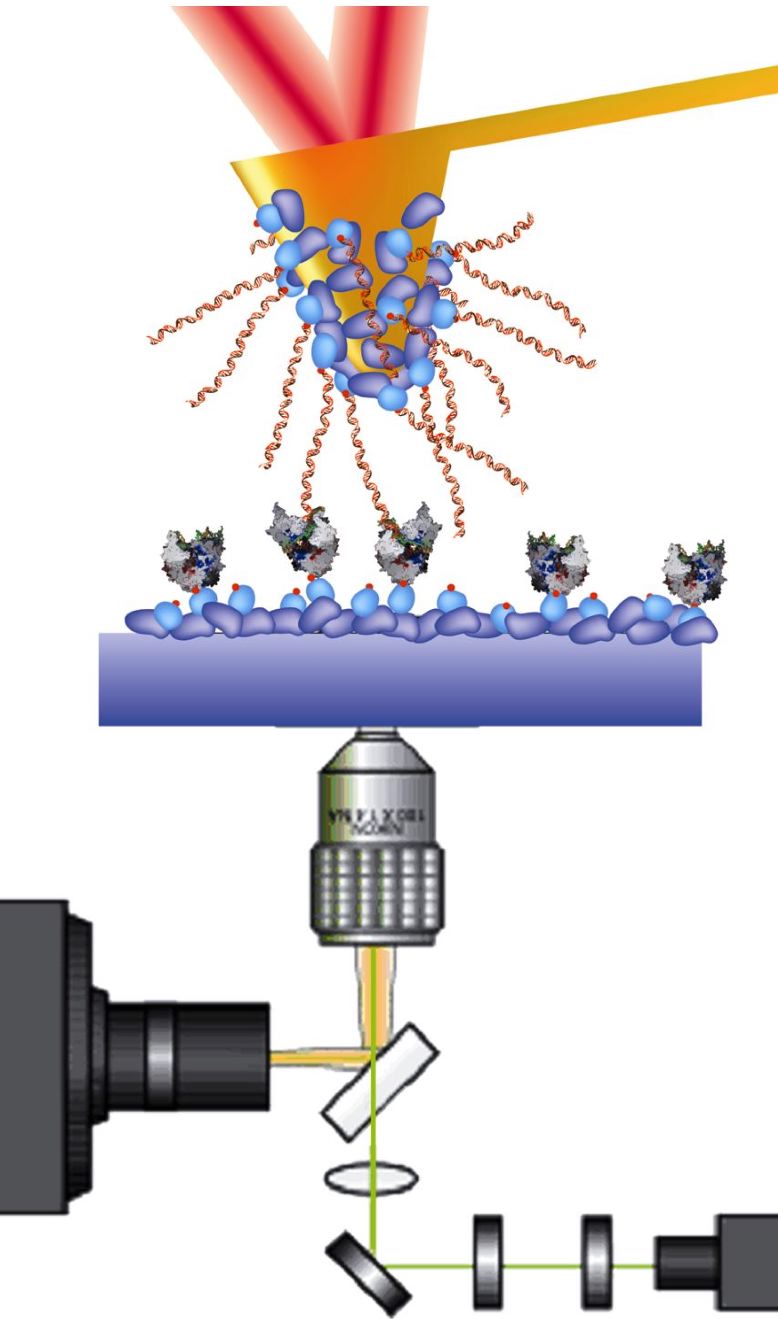
For AFM pulling experiments, we will "fish" for T7 RNAP using template DNA dangling from the AFM cantilever (see figure). Biotin-streptavidin links will fix T7 RNAP to the glass surface and also the template DNA to the cantilever. The AFM will be mounted over an inverted fluorescence microscope, and single-molecule fluorescence will be used to confirm the active transcription of T7 RNAP. Additionally, it is possible to reverse the orientation of the dangling template DNA and instead fish with a transcription-assisting pull rather than with an opposing force.
Development of a centrifugal force scanning microscope (CFSM) based on CD/DVD technology

A prototype CFSM will be further developed to facilitate single-molecule manipulation. A sealed gasket encloses the sample chamber, which will be mounted on a spinning compact disk (see figure). A ds-DNA library, containing important transcription initiation and termination sequences, will be spotted on the lower surface of the sample chamber using DNA microarray technology. Each ds-DNA, prepared with biotin at its distal end, will bind a strepavidin-coated fluorescent microsphere. The centrifugal force will push (when viewed in a rotating frame of reference) these microspheres radially and so cause the attached ds-DNA to stretch. The laser on a modified compact disk player will be used to track the bead displacement. The centrifugal force on each microsphere can be calculated based on their size, density and radial velocity. The centrifugal force plotted against microsphere displacement will provide information about the physical properties (persistence length, overstretching transition, etc) of different DNA sequences in the library. After analyzing the sequence dependence of the ds-DNA force-versus- displacement curves, the sample chamber will be filled with proteins that bind DNA: T7 RNAP binding its promoter sequences, for instance. In this example, the stretching properties of the promoter DNA will be examined; in the presence and absence of T7 RNAP (which has been shown to bend its promoter 80 degrees and unwind it by 8 base pairs). Furthermore, researchers have found that T7 RNAP responds differently to inhibition by T7 lysozyme depending on which type of promoter sequence is used to initiate transcription. CFM would be the ideal technique to quantify the interaction between T7 RNAP and all possible promoter sequences, in the presence or absence of T7 lysozyme.